Proton affinity of uracil. A computational study of protonation sites |
| |
| Authors: | Jill K. Wolken František Tureček |
| |
| Affiliation: | Department of Chemistry, University of Washington, Seattle 98195-1700, USA. |
| |
| Abstract: | 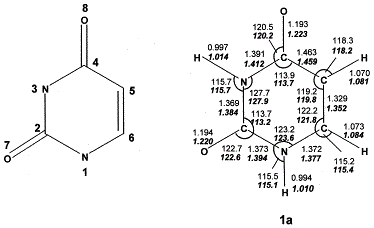
Relative stabilities of uracil tautomers and cations formed by gas-phase protonation were studied computationally with the B3LYP, MP2, QCISD, and QCISD(T) methods and with basis sets expanding from 6-31G(d,p) to 6-311+G(3df,2p). In accordance with a previous density functional theory study, the dioxo tautomer 1a was the most stable uracil isomer in the gas phase. Gibbs free energy calculations using effective QCISD(T)/6-311+G(3df,2p) energies suggested >99.9% of 1a at equilibrium at 523 K. The most stable ion isomer corresponded to N-1 protonated 2,4-dihydroxypyrimidine, which however is not formed by direct protonation of 1a. The topical proton affinities in 1a followed the order O-8 > O-7 > C-5 > N-3 > N-1. The thermodynamic proton affinity of 1a was calculated as 858 kJ mol−1 at 298 K. A revision is suggested for the current estimate included in the ion thermochemistry database. |
| |
| Keywords: | |
| 本文献已被 ScienceDirect PubMed SpringerLink 等数据库收录! |
|