Gas-phase thermochemical properties of some tri-substituted phenols: A density functional theory study |
| |
| Affiliation: | 1. Centro de Geologia da Universidade do Porto, Rua do Campo Alegre, s/n, P-4169-007 Porto, Portugal;2. Centro de Investigação em Química, Departamento de Química e Bioquímica, Faculdade de Ciências da Universidade do Porto, Rua do Campo Alegre, s/n, P-4169-007 Porto, Portugal;3. Department of Chemistry and Biochemistry, University of Maryland, Baltimore County, 1000 Hilltop Circle, Baltimore, MD 21250, USA;1. Department of Chemical Engineering, Imperial College London, SW7 2AZ, United Kingdom;2. Fluid Science & Resources Division, University of Western Australia, Crawley, WA, 6009, Australia;1. Max Planck Institute for Dynamics of Complex Technical Systems, Magdeburg, Saxony-Anhalt, Germany;2. Department of Physical Chemistry, Kazan Federal University, Kazan, Tatarstan, Russia;3. Department of Physical Chemistry and Department “Science and Technology of Life, Light and Matter,” University of Rostock, Rostock, Mecklenburg-Vorpommern, Germany |
| |
| Abstract: | 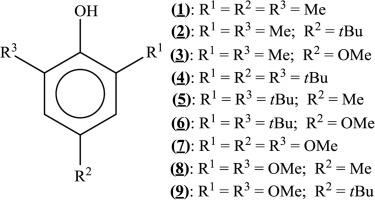
The study of the energetics of phenolic compounds has a considerable practical interest since this family of compounds includes numerous synthetic and naturally occurring antioxidants. In this work, density functional theory (DFT) has been used to investigate gas-phase thermochemical properties of the following tri-substituted phenols: 2,4,6-trimethylphenol, 2,6-dimethyl-4-tert-butylphenol, 2,6-dimethyl-4-methoxyphenol, 2,4,6-tri-tert-butylphenol, 2,6-di-tert-butyl-4-methylphenol, 2,6-di-tert-butyl-4-methoxyphenol, 2,4,6-trimethoxyphenol, 2,6-dimethoxy-4-methylphenol and 2,6-dimethoxy-4-tert-butylphenol. Molecular structures were computed with the B3LYP and the ωB97X-D functionals and the 6-31G(d) basis set. More accurate energies were obtained from single-point energy calculations with both functionals and the 6-311++G(2df,2pd) basis set. Standard enthalpies of formation of the phenolic molecules and phenoxyl radicals were derived using an appropriate homodesmotic reaction. The O H homolytic bond dissociation enthalpies, gas-phase acidities and adiabatic ionization enthalpies were also calculated. The general good agreement found between the calculated and the few existent experimental gas-phase thermochemical parameters gives confidence to the estimates concerning the phenolic compounds which were not yet experimentally studied. H homolytic bond dissociation enthalpies, gas-phase acidities and adiabatic ionization enthalpies were also calculated. The general good agreement found between the calculated and the few existent experimental gas-phase thermochemical parameters gives confidence to the estimates concerning the phenolic compounds which were not yet experimentally studied. |
| |
| Keywords: | Tri-substituted phenols Molecular structure Enthalpy of formation Acidity Adiabatic ionization enthalpy |
| 本文献已被 ScienceDirect 等数据库收录! |
|